Research objective
Determine which cell compartment shows the strongest coupling between transcription factor activity and the senescence-associated secretory phenotype (SASP) in aged human skeletal muscle, identify the transcriptional regulatory programs involved, and propose compartment-specific therapeutic strategies.
The approach:
- Donor-level Spearman correlations between transcription factor expression and a 12-gene SASP composite score (SASP12) across four compartments (vascular endothelial, FAP, MuSC, immune) in two independent single-cell atlases
- pySCENIC co-expression-module inference (GRNBoost2 + AUCell) to prioritize candidate regulators beyond mRNA-level correlation (note: cisTarget motif pruning was non-functional in this run; all module-level claims are preliminary and await motif validation)
- Partial-correlation decomposition to separate collinear signals (JUNB vs CDKN1A/p21)
- Cross-atlas integrative analysis (meta-analytic pooling, direction-of-effect voting, replication tiering)
- Vascular-to-FAP ligand-receptor crosstalk analysis for paracrine axis identification
Use the Human Lifemap Muscle Atlas (HLMA, 387,000+ cells, 23 donors) as the primary dataset, with the Nature Aging 2024 atlas (90,902 cells, 12 donors) for cross-atlas validation.
This is a living project. Marvin has finished this iteration using existing public data and literature. The next iteration begins when you contribute: a critique of the analysis, an alternative interpretation, a dataset we missed, a question we haven’t asked, or experimental validation of a finding. Marvin will incorporate your input and run the next research cycle, published openly, for free. See open questions below or learn more about how IORI works.
Summary of discoveries
-
Vascular endothelial cells show the strongest SASP coupling — Strong. JUNB co-expression-module activity correlates with SASP at rho=0.923 (p=3.64×10⁻¹⁰, N=23 donors) in vascular cells — the tightest coupling of any TF in any compartment. This is a regulatory coupling finding, not an absolute SASP burden measurement (immune cells lead on SASP-high cell fraction). Reframes the prevailing FAP-centric model of muscle aging.
-
Three-compartment regulatory model — Strong. Vascular cells run an AP-1 axis (JUNB-dominated). MuSCs run a dual AP-1 + p21 axis. FAPs operate through C/EBPβ with negative AP-1 module-level coupling — though cross-compartment module polarity may reflect tissue-specific co-expression structure rather than genuine regulatory opposition (36.8% of all TFs show zero target overlap between compartments). Each compartment requires a different therapeutic strategy.
-
KLF10 is a TGF-β bystander biomarker, not a SASP driver — Strong. Despite the strongest and most consistent mRNA-SASP correlation across five datasets (rho=0.813, I²=27.2%), module-level validation is null (AUCell rho=0.079 in FAP). Prevents a misdirected therapeutic strategy targeting KLF10 in FAPs.
-
Two distinct vascular→FAP paracrine axes — High. Axis 1: JNK-dependent inflammatory crosstalk (CXCL2→CXCR2, IL6→IL6R). Axis 2: JNK-independent vessel destabilization (ANGPT2→Tie1/Tie2). The two axes are not reducible to a single pharmacological target.
-
FAP growth factor compensation is inadequate — Moderate. FAPs upregulate FGF7 and HGF with age but the signal does not reach MuSCs: the FGF7→FGFR receptor axis is broken, and IGF2 declines across all FAP subtypes while MuSC IGF1R rises. Growth factor supplementation may bypass this failure.
-
CDK4/6 inhibitors require satellite-cell safety evaluation — Moderate. CDKN1A/p21 is consistent with a role downstream of JUNB in the regulatory cascade. CDK4/6 activity is required for MyoD-driven satellite cell re-entry. Any clinical program pairing a JNK inhibitor with a CDK4/6 inhibitor in muscle-aging populations requires satellite-cell safety screens.
-
JNK→AP-1→CDKN1A→SASP hierarchy — Moderate. Partial-correlation decomposition shows JUNB’s SASP signal is largely mediated through CDKN1A, while CDKN1A retains independent signal beyond JUNB. Consistent with CDKN1A/p21 as a pharmacodynamic biomarker for JNK-directed trials, though the ordering cannot be established from cross-sectional data alone.
-
BML-260 + ANGPT2 blockade as leading combination — Moderate. BML-260 (a DUSP22 modulator) addresses the JNK-dependent vascular inflammatory axis; ANGPT2 blockade (MEDI3617 or AKB-9778) addresses the JNK-independent vessel-destabilization axis. Neither agent alone is predicted to cover both paracrine pathways.
Open questions for the community
All findings are correlational. Causal validation requires perturbation in primary human muscle cell cultures, not mouse models (the AP-1 polarity pattern was not conserved in a cross-species comparison with Tabula Muris Senis, though protocol differences preclude definitive species-comparison). We are actively looking for:
- JNK inhibition in primary human vascular endothelial cells. JNK must be targeted at the kinase/phosphatase layer (JNK engages JUNB post-translationally; siJUNB or CRISPRi would not recapitulate a JNK-inhibitor phenotype). BML-260 is the lead candidate but has not been tested in aged endothelial cells specifically.
- C/EBPβ perturbation in primary human FAPs. The FAP SASP mechanism operates through a fundamentally different TF program (C/EBPβ, not AP-1). Module-level confirmation is currently underpowered (AUCell rho=0.474, p=0.064 at N=16 snRNA-filtered donors).
- A third independent vascular muscle aging atlas. Cross-atlas replication is directionally concordant but formally underpowered (Nature Aging endothelial rho=0.720, p=0.008, N=12 donors, observed power 0.753 vs 80%-power floor of 0.745). No fully independent vascular muscle aging atlas currently exists.
- FGF7/HGF/IGF2 supplementation studies in MuSC activation assays. The broken FAP→MuSC growth factor axis predicts that exogenous supplementation should bypass the endogenous signaling failure. FGF7 (palifermin) is already FDA-approved for oral mucositis. Note: FGF7 may signal through FGFR2 rather than FGFR1 in satellite cells — receptor specificity should be verified.
- CDK4/6 + JNK inhibitor satellite-cell safety screens. The safety concern is inferred from the p21 hierarchy and established satellite-cell biology, not directly tested in the context of muscle aging.
- Human iPSC-derived vascular organoid models. Cross-species re-analysis shows the AP-1 polarity pattern may not be conserved in mouse (Tabula Muris Senis: 1 of 5 directional matches in EC, 2 of 5 in FAP). Standard preclinical mouse models may not recapitulate these axes.
If you have data, capacity, or relevant perturbation systems, file an issue on the project repository or email [email protected].
Findings
Vascular JUNB-SASP dominance
Among 21 candidate TFs spanning AP-1, KLF, EGR, IRF, C/EBP, NF-κB, and STAT families, JUNB ranked first in vascular endothelial cells with rho=0.929 (p=1.65×10⁻¹⁰, N=23 donors). CDKN1A (p21) ranked second (rho=0.913), followed by EGR1 (rho=0.908). All seven Jun/Fos subunits showed rho > 0.84, indicating a coherent AP-1 regulatory axis rather than a single TF effect.
pySCENIC co-expression-module inference (GRNBoost2 + AUCell; motif validation pending due to cisTarget technical failure) supports JUNB as the leading candidate regulator: JUNB module AUCell scores correlated with SASP12 at rho=0.923 (p=3.64×10⁻¹⁰), nearly identical to the raw mRNA correlation (delta rho = +0.002). This module-level agreement prioritizes JUNB beyond mere mRNA-level biomarker status, though motif-validated regulatory confirmation remains pending.
The JNK pathway showed a distinctive post-translational activation pattern: upstream kinase mRNAs (MAP3K, MAP2K4/7, MAPK8/9/10) had null age effects (Cohen’s d ≈ 0) while downstream immediate-early AP-1 targets (FOS, JUNB) were robustly age-upregulated (FOS Cohen’s d=0.907, JUNB d=0.901 in vascular cells). This is consistent with JNK activation by phosphorylation rather than transcriptional upregulation of the kinase cascade.
The JUNB-SASP coupling survived multiple verification tests: IL6⁺ venular exclusion (rho changed from 0.9262 to 0.9287), leave-one-out analysis (minimum rho=0.9185), senescence-marker comparison (JUNB correlated more strongly with CDKN1A/p21 than with endothelial activation markers ICAM1, VCAM1, or SELE), and cross-atlas replication in Nature Aging endothelial cells (rho=0.720, p=0.008, N=12, directionally concordant but formally underpowered). Country of origin (China vs Spain) was the dominant confound in HLMA, not age (rho(JUNB, country)=0.626 vs rho(JUNB, age)=0.042); within-China stratification confirmed the coupling survives (rho=0.947, N=14). Within-country analysis confirmed JUNB expression is elevated in aged versus young donors in both population strata (China Mann-Whitney p=0.029; Spain directionally consistent, underpowered at N=9).
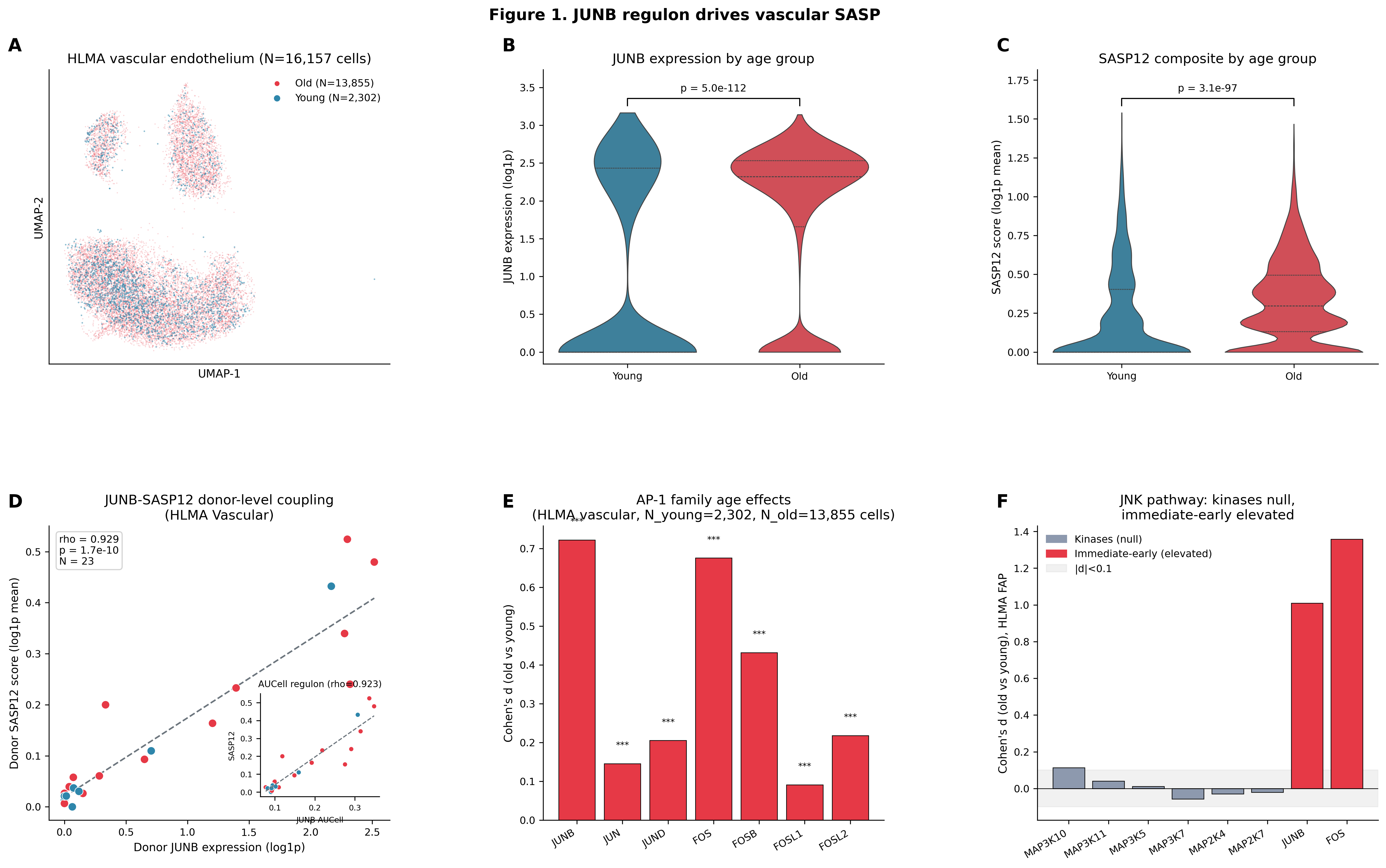 Figure 1. JUNB/AP-1 co-expression-module activity is tightly coupled to vascular SASP at the donor level. (A) UMAP of vascular endothelial cells by age. (B) JUNB expression by age group. (C) SASP12 composite by age group. (D) Donor-level JUNB-SASP scatter (rho=0.929, N=23). Inset: AUCell module activity vs SASP (rho=0.923). (E) AP-1 family age effects. (F) JNK kinase cascade: null upstream, elevated downstream.
Figure 1. JUNB/AP-1 co-expression-module activity is tightly coupled to vascular SASP at the donor level. (A) UMAP of vascular endothelial cells by age. (B) JUNB expression by age group. (C) SASP12 composite by age group. (D) Donor-level JUNB-SASP scatter (rho=0.929, N=23). Inset: AUCell module activity vs SASP (rho=0.923). (E) AP-1 family age effects. (F) JNK kinase cascade: null upstream, elevated downstream.
Confidence: Strong. Meets all five pre-registered evidence criteria. The single limitation is absence of a fully independent third vascular muscle aging atlas.
Three-compartment TF architecture
The TF-SASP coupling landscape differs dramatically across compartments:
- Vascular: AP-1 axis. JUNB rho=0.929, CDKN1A rho=0.913, EGR1 rho=0.908.
- MuSC: Dual AP-1 + p21 axis. CDKN1A rho=0.929, JUNB rho=0.934, EGR1 rho=0.917. The p21 axis is the dominant distinguishing feature (donor-level JUNB-p21 collinearity rho=0.944).
- FAP: C/EBP-EGR-KLF axis. CEBPB rho=0.802, EGR2 rho=0.799, KLF10 rho=0.784. JUNB is last at rho=-0.014. All seven canonical AP-1 subunits show negative AUCell module coupling in FAPs — but cross-compartment co-expression modules share near-zero target-gene overlap (36.8% of all TFs have Jaccard=0.000 between vascular and MuSC), so this polarity reversal may reflect tissue-specific co-expression structure rather than genuine regulatory opposition. AP-1-directed interventions in FAPs require direct perturbational testing.
- Subtype-restricted analysis confirmed that RUNX2+ FAPs — the subtype most enriched for JUNB-positive cells by binary classification (4.37×) — actually have the weakest continuous JUNB-SASP coupling of any FAP subtype (rho=0.086), ruling out a masked subpopulation explanation for the donor-level null.
After joint partial-correlation adjustment for age, sex, and sequencing technology, the hierarchy sharpens: vascular JUNB partial rho=0.912 (N=23); FAP CEBPB partial rho=0.888 (N=22, strengthened by +0.066); MuSC EGR1 partial rho=0.622 (N=23, JUNB demoted to second place after confounder adjustment).
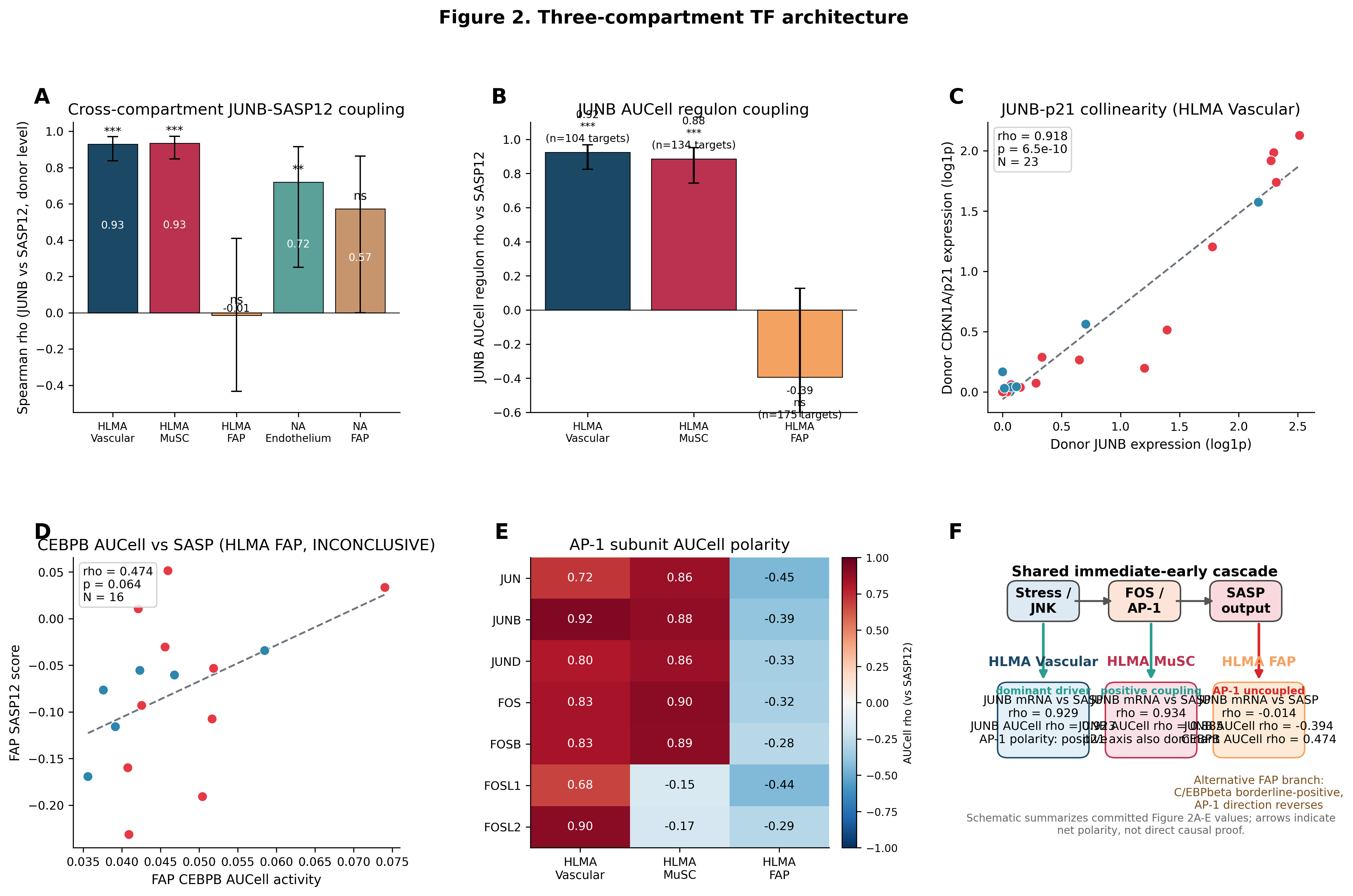 Figure 2. Three-compartment TF-SASP architecture. (A) Cross-compartment JUNB-SASP at donor level. (B) JUNB AUCell module coupling by compartment. (C) JUNB-CDKN1A collinearity. (D) FAP CEBPB AUCell. (E) AP-1 polarity heatmap — all seven subunits negative in FAP. (F) Schematic model.
Figure 2. Three-compartment TF-SASP architecture. (A) Cross-compartment JUNB-SASP at donor level. (B) JUNB AUCell module coupling by compartment. (C) JUNB-CDKN1A collinearity. (D) FAP CEBPB AUCell. (E) AP-1 polarity heatmap — all seven subunits negative in FAP. (F) Schematic model.
Confidence: Strong for the vascular and FAP programs. High for MuSC (cross-atlas CDKN1A coupling present but below the rho>0.5 effect-size threshold).
KLF10 bystander
KLF10 showed the strongest mRNA-level SASP coupling of any TF in FAPs (rho=0.862, p<0.001, N=22 donors) and was the only TF positive in all five dataset-compartment combinations at the rho>0.5 threshold (cross-dataset I²=27.2%, the lowest heterogeneity of any TF).
However, pySCENIC AUCell analysis argues against KLF10 as a SASP driver. KLF10 AUCell rho=-0.164 (p=0.455) versus raw mRNA rho=0.836 (delta=-1.000) in HLMA vascular cells. In HLMA FAPs, KLF10 AUCell rho=0.079 (p=0.770) versus raw mRNA rho=0.862 (delta=-0.782). KLF10’s co-expression module showed near-zero or negative association with SASP despite strong mRNA-level correlation.
The mechanistic explanation: KLF10 (TIEG1) is a TGF-β early response gene and transcriptional repressor that recruits Sin3A/HDAC complexes. In aging muscle, TGF-β signaling activates both KLF10 mRNA (as a negative feedback regulator) and SASP genes (via SMAD-independent branches including AP-1). KLF10 mRNA rises with SASP because both respond to the same upstream TGF-β signal, but KLF10’s repressive targets are distinct from SASP output genes. The KLF10 co-expression module in vascular cells (74 targets) contained zero SASP genes, compared to JUNB’s 104-target module which included CXCL2 and CCL2.
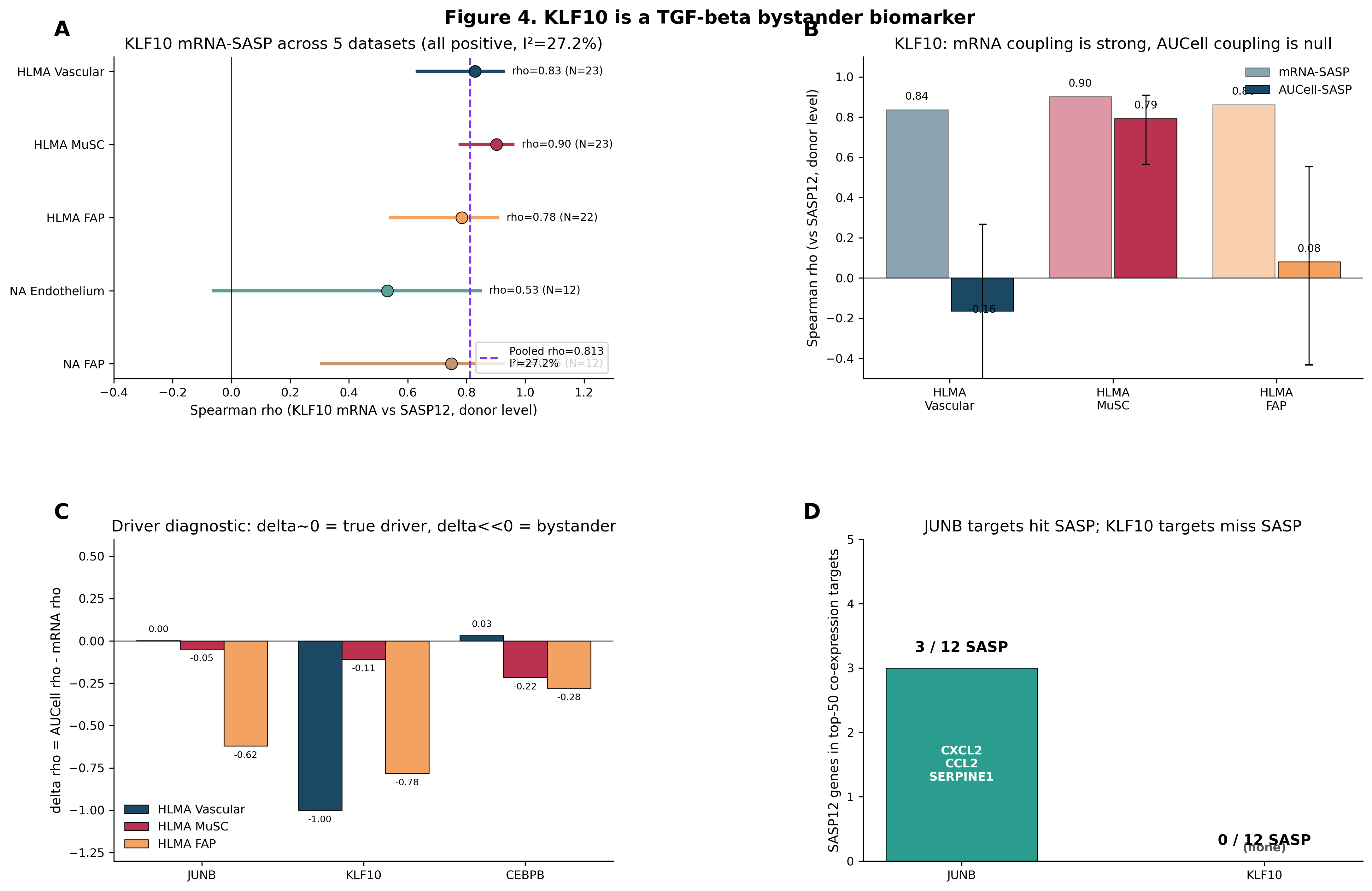 Figure 4. KLF10 is a TGF-β-responsive bystander biomarker. (A) KLF10 mRNA-SASP across 5 datasets (all positive). (B) KLF10 AUCell-SASP (near-zero). (C) Delta diagnostic: JUNB delta≈0 (candidate driver), KLF10 delta≈-1.0 (bystander). (D) Target-gene overlap with SASP: JUNB includes CXCL2/CCL2; KLF10 has zero SASP genes.
Figure 4. KLF10 is a TGF-β-responsive bystander biomarker. (A) KLF10 mRNA-SASP across 5 datasets (all positive). (B) KLF10 AUCell-SASP (near-zero). (C) Delta diagnostic: JUNB delta≈0 (candidate driver), KLF10 delta≈-1.0 (bystander). (D) Target-gene overlap with SASP: JUNB includes CXCL2/CCL2; KLF10 has zero SASP genes.
Confidence: Strong as a negative result. Prevents a misdirected therapeutic strategy targeting KLF10 in FAPs.
Vascular-FAP crosstalk
Donor-level Spearman coupling between 49 vascular ligand-FAP receptor pairs resolved two mechanistically distinct paracrine axes:
Axis 1: JNK-dependent inflammatory crosstalk. Four pairs showed strong positive vascular-JUNB coupling: CXCL2→CXCR2 (rho_JUNB=0.924, BH-FDR q=1.5×10⁻⁸), IL6→IL6R (rho_JUNB=0.855), TNF→TNFRSF1A (rho_JUNB=0.824), VEGFA→KDR (rho_JUNB=0.766). CXCL2→CXCR2 and VEGFA→KDR survive 49-pair BH-FDR. Because vascular JUNB activity is the presumed JNK readout, these ligand outputs are predicted to be reducible by JNK inhibition.
Axis 2: JNK-independent vessel destabilization. ANGPT2→TIE1 (and ANGPT2→TEK) showed moderate coupling to donor age (rho_age=+0.465, p=0.025) and to vascular JUNB (rho_JUNB=0.442, p=0.035). Because ANGPT2 mean expression is not a JNK-downstream AP-1 target in our data, this axis is predicted to persist under JNK-targeted therapy and requires a mechanistically distinct intervention. Two Tie2-axis therapeutic candidates (MEDI3617, a neutralizing anti-ANGPT2 monoclonal; AKB-9778/razuprotafib, a VE-PTP inhibitor) converge on Tie2 activation but have not been tested jointly in aged-muscle cohorts.
Three additional pairs showed age-associated loss of protective paracrine factors: SEMA3C→NRP1 (rho_age=-0.550), SEMA3F→NRP2 (rho_age=-0.500), FGF1→FGFR1 (rho_age=-0.490). These did not couple to vascular JUNB and did not survive 49-pair multiple-testing correction. JNK inhibition is not expected to restore these ligands; restoration strategies (recombinant FGF1, VEGFB mimetics) would be required separately.
Therapeutic implication: Vascular SASP → FAP signaling is not a single pharmacological entity. JNK inhibition (Axis 1) plus ANGPT2/Tie2 stabilization (Axis 2) are complementary; addressing only Axis 1 leaves the vessel-destabilization and protective-loss programs intact.
Confidence: High. Axis 1 survives 49-pair BH-FDR (2 of 4 pairs). Axis 2 and protective-factor loss do not survive correction and are classified as Moderate.
FAP growth factor compensation
Despite their distinct SASP regulatory architecture, FAPs showed a regenerative-compensatory secretome profile at the population level. Aged FAPs upregulated FGF7 (Cohen’s d=+1.29) and HGF (d=+1.10) while decreasing pro-inflammatory ligands TNF (d=-0.54) and IL6 (d=-0.51). This pattern is regenerative-compensatory, not pro-inflammatory.
However, this molecular compensation did not translate to functional benefit. The FAP growth factor score did not predict MuSC activation at the donor level (rho=0.189, p=0.41, N=21 matched donors). The FGF7→FGFR crosstalk axis was broken: FAPs surged FGF7 (d=+1.29) but MuSCs did not upregulate the relevant receptor (d=+0.13, flat). IGF2 declined across all FAP subtypes (d=-0.60 to -1.24) while MuSCs upregulated IGF1R (d=+0.89), creating a “ligand-receptor mismatch” where MuSCs are primed to receive growth signals that are not being sent.
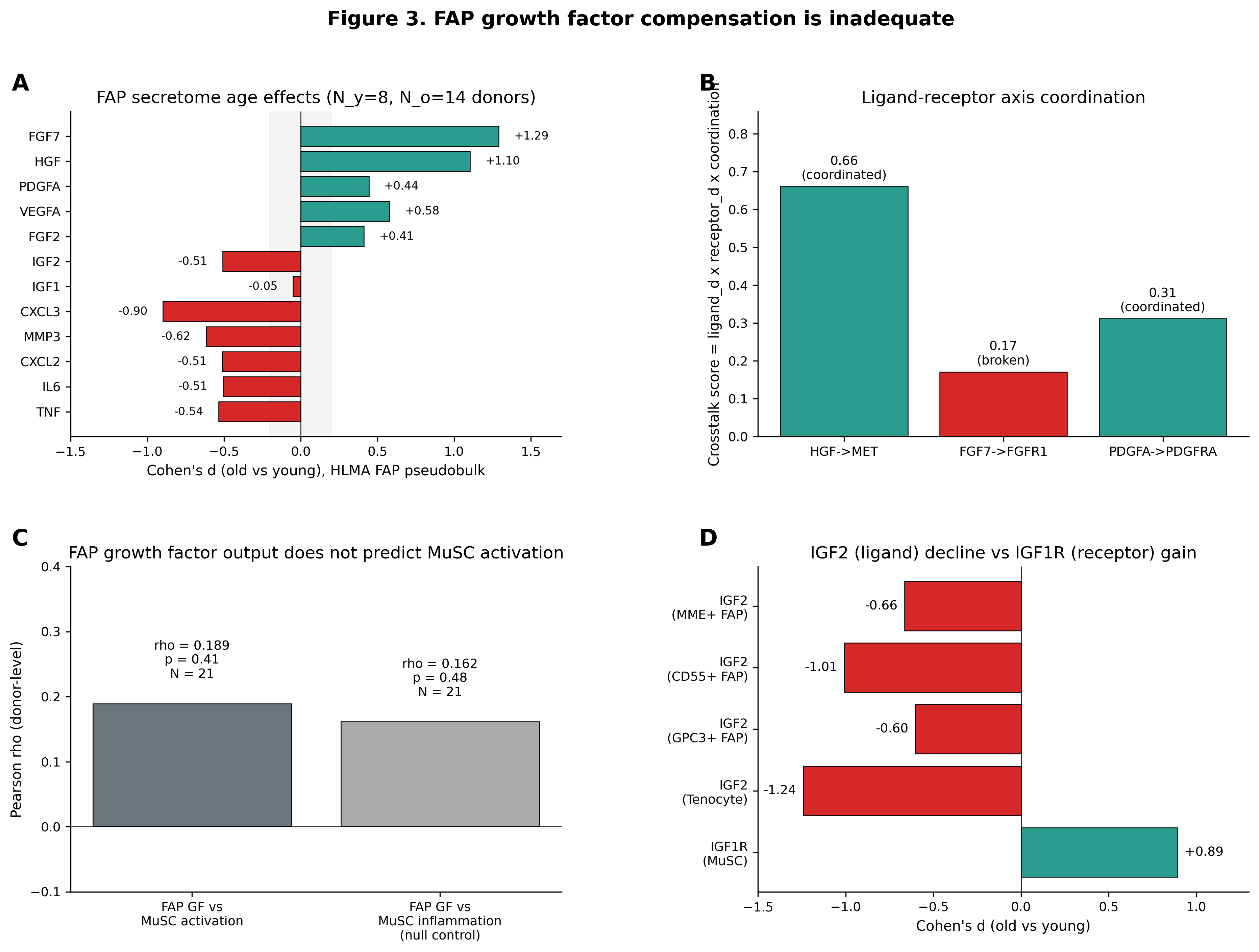 Figure 3. FAP growth factor compensation is inadequate. (A) FAP secretome age effects. (B) Crosstalk axis coordination scores. (C) FAP growth factor score vs MuSC activation (NS). (D) IGF2 decline vs IGF1R upregulation.
Figure 3. FAP growth factor compensation is inadequate. (A) FAP secretome age effects. (B) Crosstalk axis coordination scores. (C) FAP growth factor score vs MuSC activation (NS). (D) IGF2 decline vs IGF1R upregulation.
Exogenous growth factor supplementation (FGF7/KGF, HGF, IGF1/IGF2) is justified to bypass the broken endogenous FAP→MuSC signaling axis. FGF7 (palifermin) is FDA-approved for oral mucositis, providing a potential repurposing pathway. FGF7 is reported to act through FGFR2 rather than FGFR1 in satellite cells, which may modify interpretation of this axis.
Confidence: Moderate. Growth factor compensation is clearly documented; the broken crosstalk axis is consistent but based on single-atlas population-level effects.
CDK4/6 safety
The CDKN1A/p21-SASP coupling (donor-level rho=0.929, p=1.66×10⁻¹⁰, N=23 in MuSC) operates in parallel with AP-1. CDK4/6 activity is required for MyoD-driven cell-cycle re-entry in satellite cells. Pharmacological CDK4/6 inhibition establishes a p53-dependent senescent state with a restricted secretory phenotype, consistent with a senomorphic action of palbociclib that is distinct from its impact on muscle stem cell activation.
CDKN1A/p21 is a candidate pharmacodynamic biomarker for JNK-directed trials, not a therapeutic target in the MuSC compartment. Any clinical program that pairs a JNK inhibitor with a CDK4/6 inhibitor in muscle-aging populations requires satellite-cell safety screens as a prerequisite.
Confidence: Moderate. The safety concern is inferred from established satellite-cell biology and the p21 hierarchy; the supporting evidence is from literature synthesis, not direct experimentation in this study.
JNK-AP1-CDKN1A hierarchy
Partial-correlation decomposition on donor-level scores in HLMA vascular cells: raw rho(JUNB, SASP)=0.937, rho(CDKN1A, SASP)=0.935, rho(JUNB, CDKN1A)=0.927. Partialing out CDKN1A largely extinguishes the JUNB signal (partial rho(JUNB|SASP|CDKN1A)=0.137, p=0.531, 95% CI [−0.335, 0.636]), whereas partialing out JUNB preserves a substantial CDKN1A signal (partial rho(CDKN1A|SASP|JUNB)=0.513, p=0.012).
This is consistent with a model in which JUNB’s SASP effect is largely mediated through CDKN1A, while CDKN1A retains an independent SASP-associated signal beyond JUNB. However, the partial correlations are in the multicollinearity regime (rho(JUNB, CDKN1A)=0.927), and the ordering cannot be established from cross-sectional data. If this model holds, CDKN1A/p21 would be the more proximal pharmacodynamic readout for JNK-directed trials than JUNB mRNA.
Three independent genetic-regulation layers converge on the conclusion that AP-1 immediate-early TFs are not transcriptionally regulated at the genomic level: upstream kinase mRNAs show null age effects, bulk muscle eQTL from Open Targets yields 0 of 54 TF×sarcopenia/lean-body-mass trait colocalizations, and single-cell eQTL in OneK1K (N=982) finds JUNB, FOS, EGR1, and ATF3 null at the cis-eQTL level in immune cells, while CDKN1A carries a significant cis-eQTL (p=3.4×10⁻¹⁰). The therapeutic entry point is the JNK/MAPK kinase layer, not the TF-DNA interface.
Confidence: Moderate. HLMA-only; causal direction is inferred from post-translational literature, not directly tested.
Therapeutic model
Based on the three-compartment model and two-axis paracrine architecture, the leading combination hypothesis:
Vascular, Axis 1 (JNK-dependent). BML-260, a preclinical DUSP22 modulator that represses JNK activity and ameliorates skeletal muscle wasting in human-muscle-cell, sarcopenia, glucocorticoid, and ICU-catabolism models. Unlike siJUNB/CRISPRi, it targets the kinase/phosphatase layer at which JNK engages JUNB post-translationally. First-generation systemic JNK inhibitors encountered translational bottlenecks: tanzisertib (CC-930) was discontinued in phase 2 for liver enzyme elevations, and CC-90001 failed its IPF phase 2 efficacy endpoint (terminated by sponsor decision).
Vascular, Axis 2 (JNK-independent vessel destabilization). Tie2-axis therapeutic candidates MEDI3617 (neutralizing anti-ANGPT2 monoclonal) and AKB-9778/razuprotafib (VE-PTP inhibitor) converge on Tie2 activation. This axis is predicted to persist under BML-260 monotherapy because ANGPT2 is not downstream of vascular JUNB in our data.
MuSCs. CDK4/6 inhibitors require safety evaluation before clinical use in muscle-aging populations. CDKN1A/p21 is a candidate biomarker, not a target. The relevant therapeutic strategy is to protect satellite-cell activation capacity, not to suppress the CDKN1A axis.
FAPs. AP-1-directed interventions in FAPs require direct testing before therapeutic conclusions; the negative module-level association may reflect tissue-specific co-expression structure rather than regulatory opposition. C/EBPβ is the leading candidate regulator. FAPs are the dominant senolytic target in muscle (navitoclax-sensitive, BCL-2-dependent), and senolytic therapy targeting FAPs is complementary to, not competing with, JNK-directed therapy of the vascular compartment.
The BML-260 + ANGPT2-blockade combination is our leading hypothesis; neither agent alone is expected to address the other axis.
Confidence: Moderate. The combination is inferred from the compartment-specific paracrine architecture; none of the agents have been tested jointly in aged-muscle cohorts.
Sources, datasets, and literature
Datasets
- HLMA atlas (CNGBdb OMIX004308, Lai et al.) — 387,000+ cells/nuclei from donors aged 15-99 years. Compartment-specific files: Vascular (N=23 donors, 16,157 cells after IL6⁺ exclusion), MuSC (N=23 donors, 9,559 cells), FAP (N=22 donors, 40,389 cells), Immune (N=12 donors, 13,773 cells, snRNA-only subset).
- Nature Aging 2024 atlas (Kedlian et al., Sanger cellxgene) — 90,902 nuclei from 17 donors. Fibroblast file (N=12 donors with fibroblast subtypes) and endothelial cells (N=12 donors).
- GTEx v8 bulk muscle (N=803) — Used for post-mortem ischemic-time confound triangulation (SMTSISCH covariate). Three independent ischemic-confound failures documented; ischemic-time gating proposed as prerequisite for future bulk-muscle aging analyses.
- OneK1K single-cell eQTL (N=982, 14 immune cell types) — cis-eQTL validation for AP-1 TFs vs CDKN1A.
- Open Targets / Genetics Portal — Bulk muscle eQTL colocalizations (54 TF×sarcopenia/lean-body-mass trait combinations, 0 hits).
Software
pySCENIC 0.12.x (GRNBoost2 + AUCell; cisTarget motif pruning non-functional), Python (scipy, scanpy, pingouin, gseapy), R (partial correlation, Fisher-z meta-analysis).
Selected literature
- Lee SH et al. 2025. Modulating phosphatase DUSP22 with BML-260 ameliorates skeletal muscle wasting via Akt-independent JNK-FOXO3a repression. EMBO Mol Med. doi:10.1038/s44321-025-00234-2.
- Li Y et al. 2025. Multiomics and cellular senescence profiling of aging human skeletal muscle uncovers Maraviroc as a senotherapeutic approach for sarcopenia. Nat Commun 16:6207. doi:10.1038/s41467-025-61403-y.
- Wang B et al. 2022. Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 41(6):e108946. doi:10.15252/embj.2021108946.
- Kedlian VR et al. 2024. Human skeletal muscle aging atlas. Nat Aging. 4(5):727-744. doi:10.1038/s43587-024-00613-3.
- Lai Y et al. 2024. Multimodal cell atlas of the ageing human skeletal muscle. Nature. 629(8010):154-164. doi:10.1038/s41586-024-07348-6.
Full corpus, including rejected sources and iteration audit trail, in the project repository.